
DCB產資組ITIS研究團隊
鄭宇婷
2025年4月
美國為全球最大的藥品市場,也是新藥廠商的必爭之地。因此,該國的藥品監管單位所核准之新藥對產業而言極具重要性,由美國核准之藥品數量、類型及適應症可以觀察產業趨勢、技術發展及市場競爭格局,因此,透過觀察其所核准之新藥,做為產業趨勢的風向球,亦藉此挖掘投資機會。本文將分析2024年美國食品暨藥物管理局(Food and Drug Administration, FDA)核准之藥品概況,並針對重要產品進行介紹。
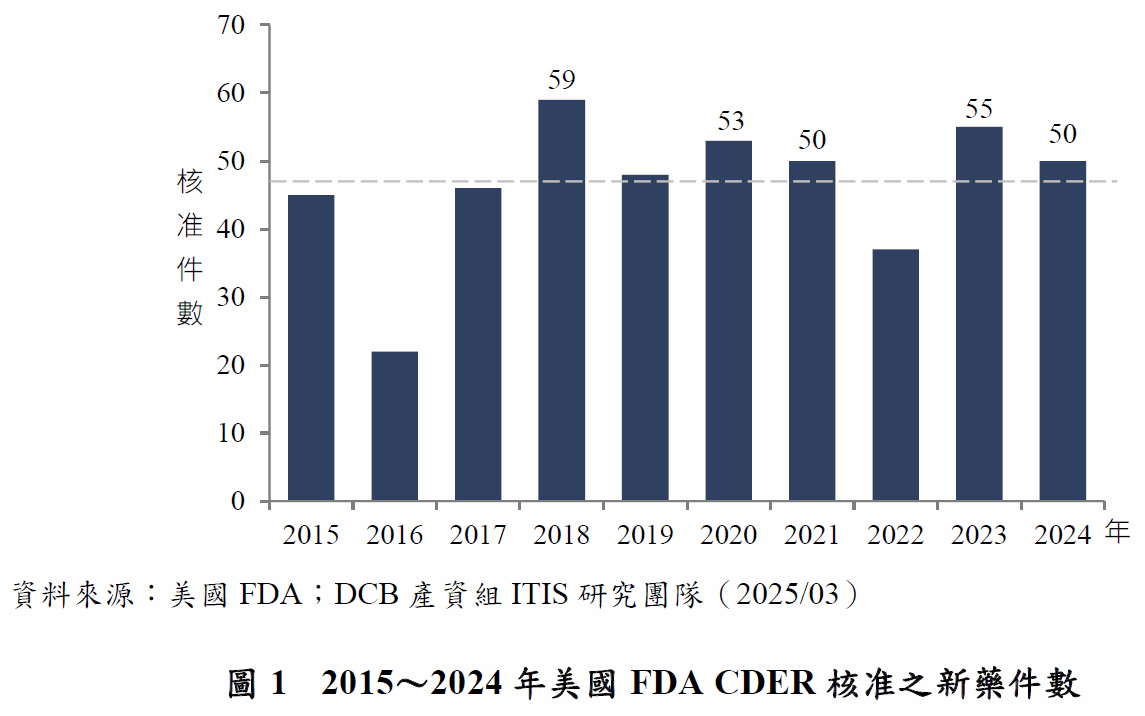
一、2024年CDER核准新藥件數達50件,高於10年平均值
2024年美國FDA的藥物評估和研究中心(Center for Drug Evaluation and Research, CDER)年核准50項新藥,核准數量略低於2023年,但仍在10年核准新藥件數的平均(47項)之上(圖1)。2024年生物製劑評估和研究中心(Center for Biologic Evaluation and Research, CBER)核准之生物製劑產品數量則有17項,其中包括mRNA疫苗、細胞治療產品、基因治療產品和血液製劑等。
分析2024年新藥取得CDER核准類型,其中34項為新分子實體(New Molecular Entities, NME),16項則為生物製劑許可申請(Biologics License Applications, BLA)(圖2)。另再細分其藥品類型,NME中30項為小分子藥品,2項為核酸藥品,1項為放射性診斷藥物,1項為胜肽藥品;BLA則是有10項為單株抗體,3項為雙特異性抗體,2項為項融合蛋白,1項為類毒素。
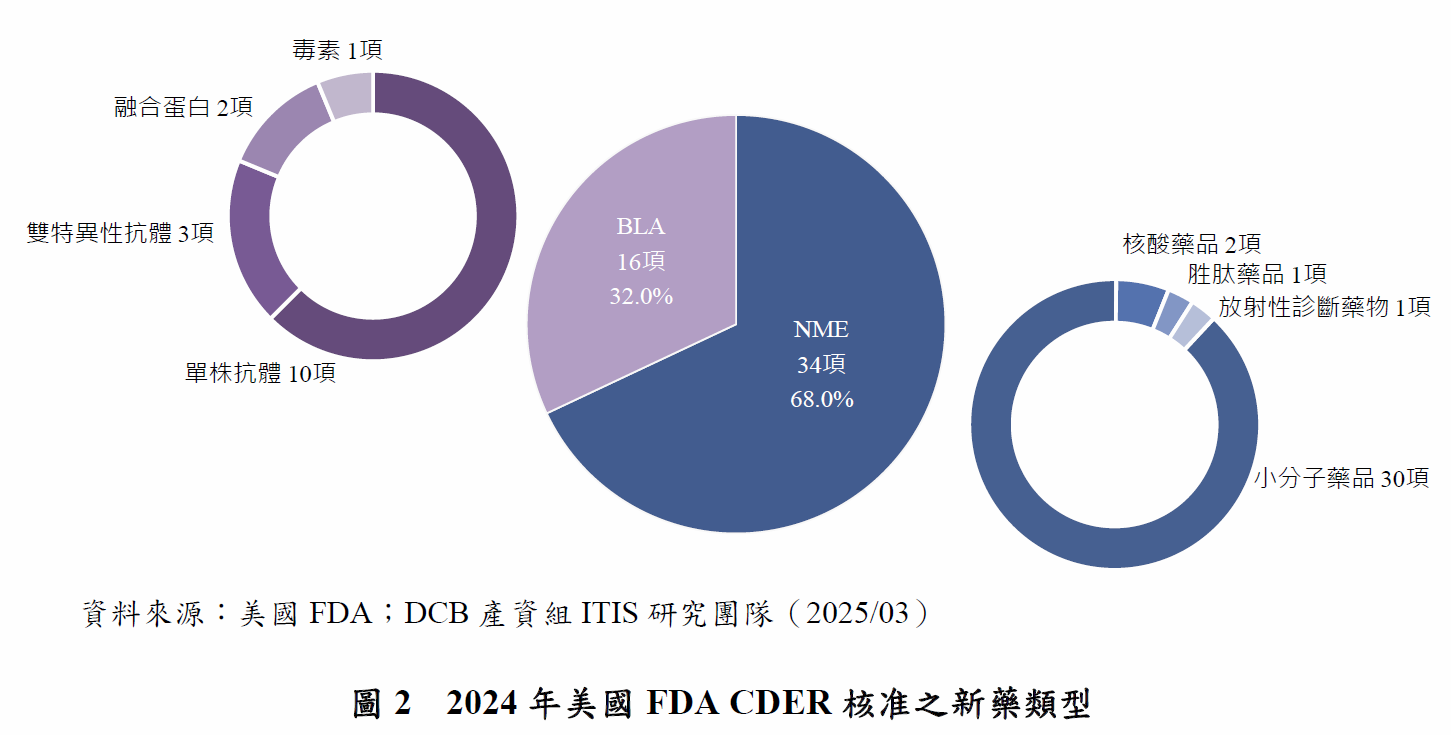
為使患者可以更快使用到新藥治療疾病,美國FDA使用多種機制以加速新藥上市,包括處方藥使用者付費法案(Prescription Drug User Fee Act, PDUFA)及針對嚴重疾病的加速計畫。其中,PDUFA主要目的之一就是透過收取藥廠的費用,增加FDA的審查人力與資源,進而縮短新藥的審查時間,加快藥品上市的速度,根據PDUFA目標,新藥審查的期限為接受藥品申請後10個月,若新藥取得優先指定審查,則為6個月。2024年核准的新藥中,有94%(47項)達到CDER的PDUFA目標日期,74%(37項)為審查第一週期即獲核准[1]。而針對嚴重疾病的加速計畫包括快速通道(fast track designation)、突破性療法(breakthrough therapy designation)、優先審查指定(priority review designation)及加速審批(accelerated approval),2024年取得CDER上市許可的產品則有66%(33項)使用其中一項或多項的加速計畫,這協助推進新藥更快速的進入市場。根據統計,2024年CDER核准的新藥當中,有68%(34項)為全球首次在美國獲准上市,顯示這些策略有助於吸引及加速廠商推動產品進入美國市場。
二、2024年多項創新機制或新興類型藥品取得上市許可
2024年美國FDA核准的新藥當中,有多項具創新機制的重要新藥上市,滿足尚未被滿足的醫療需求。例如Karuna Therapeutics的精神分裂症藥品Cobenfy(xanomeline及trospium chloride),ImmunityBio的膀胱癌突破性新藥Anktiva(nogapendekin alfa inbakicept),Verona Pharma的成人慢性阻塞性肺病(Chronic Obstructive Pulmonary Disease, COPD)治療藥品Ohtuvayre(ensifentrine),Madrigal Pharmaceuticals 的非酒精性脂肪性肝炎(Non-Alcoholic Steatohepatitis, NASH)(現已更名為代謝功能障礙相關脂肪性肝炎(Metabolic Dysfunction-Associated Steatohepatitis, MASH),後以MASH代稱)藥品Rezdiffra(resmetirom)等。
此外,創新技術突破,推動新興藥品類型進入市場,如Iovance Biotherapeutics腫瘤浸潤淋巴細胞(Tumour-Infiltrating Lymphocyte, TIL)療法Amtagvi(lifileucel)、Adaptimmune的T細胞受體(T-cell receptor, TCR)-T細胞療法Tecelra(afamitresgene autoleucel)。以下分別介紹。
1. 精神分裂症治療藥品 Cobenfy
神經/精神病學領域的藥品在2024年取得了新進展,BMS的M1/M4毒蕈鹼乙醯膽鹼受體調節劑(M1/M4 muscarinic Acetylcholine Receptor modulator, mAChR)Cobenfy,為小分子藥品,被核准用於治療精神分裂症。Cobenfy由xanomeline及trospium chloride組成,2012年Karuna Therapeutics自Eli Lilly取得xanomeline授權,並投入開發,2023年底BMS則以140億美元收購該公司,並取得Cobenfy。Eli Lilly放棄xanomeline的原因在於雖然能有效改善認知問題,但是副作用十分明顯,包括腹瀉、出汗和唾液分泌過多等,在臨床II期試驗中,超過50%接受較高劑量的患者退出臨床試驗,雖Eli Lilly及其他公司嘗試增加藥品的選擇性以降低副作用,但副作用影響仍大,因此最終Eli Lilly以僅10萬美元的價格將其授權給Karuna Therapeutics。
Karuna Therapeutics研究xanomeline之作用機制,發現xanomeline同時活化腦內及腦外之M1/M4 mAChR,其中腦外M1/M4 mAChR的活化造成副作用。為改善副作用,Karuna Therapeutics結合xanomeline及trospium chloride,其中xanomeline活化腦內的M1/M4 mAChR以達到治療效果,trospium chloride則是抑制腦外M1/M4 mAChR,降低副作用。也因為Cobenfy的臨床療效、安全性及耐受性結果表現佳,因其為新作用之機制藥物,有機會可用於對現有藥品反應不佳的患者,故市場對其抱持期待,預估2030年銷售額可望超過33億美元。
2. 膀胱癌治療藥品 Anktiva
癌症仍為2024年美國FDA核准的重要治療領域,多項重要的藥品取得美國FDA上市許可,其中,ImmunityBio的膀胱癌突破性新藥IL-15受體促進劑Anktiva,是一種融合蛋白,將可活化自然殺手細胞、CD8+殺手T細胞、樹突細胞,為多重影響之免疫療法。2023年5月美國FDA拒絕ImmunityBio的BLA,因在對第三方製造商進行許可前檢查期間記錄的問題,至2023年10月ImmunityBio重提申請,才於2024年取得上市許可。
Anktiva與卡介苗(Bacillus Calmette-Guérin, BCG)疫苗聯合治療對BCG疫苗治療無反應的非肌肉浸潤性膀胱癌(Non-Muscle Invasive Bladder Cancer, NMIBC)原位癌,治療效果表現亮眼。膀胱癌是全球第10大最常見癌症,其中約80%的病例為NMIBC。目前標準治療為膀胱內滴注BCG疫苗,但約30~40%的患者使用該治療方式無效,且最初治療反應良好的患者仍有一半的復發風險。Anktiva透過活化自然殺手細胞、CD8+殺手T細胞、樹突細胞,誘導腫瘤細胞死亡,進而達到治療效果。臨床試驗結果顯示,Anktiva與BCG疫苗合併治療可顯著提高完全緩解率(Complete Response, CR)達到62%,Keytruda的CR則為47%。
Anktiva的定價為每劑35,800美元,該價格並未包括BCG疫苗的費用。後續ImmunityBio業已規劃向美國FDA提出申請擴充Anktiva適應症,預估未來市場可望成長,但也意味著BCG疫苗的需求亦將成長。為確保BCG疫苗的供應無虞,2024年5月,ImmunityBio與印度血清研究所合作,將由印度血清研究所生產供應BCG疫苗,避免病患使用Anktiva治療時,卻發生BCG疫苗缺貨的狀況,預計在2025年第一季提交替代來源之申請。
3. 成人慢性阻塞性肺病(COPD)治療藥品 Ohtuvayre
目前針對COPD並無有效療法,無法治癒,目前僅能用藥控制疾病症狀,多採用類固醇類藥物,但有許多副作用。由Verona Pharma所開發的Ohtuvayre,採吸入式療法,作為成人COPD的維持治療使用,為20多年來第1個治療COPD的新機制藥品。透過抑制磷酸二酯酶3及4(Phosphodiesterase 3及Phosphodiesterase 4, PDE3及PDE4)雙靶點,達到支氣管擴張及抗發炎作用。
Ohtuvayre於2024年6月取得上市許可後,於第3季開始供貨。其用藥方式為每天噴兩劑,每月藥品費用為2,950美元,預估一年藥費為35,400美元。2024年Ohtuvayre的銷售額為4,200萬美元,其中第4季銷售額為3,600萬美元。在處方量的部分,全年取得16,000個訂單,其中約1/3為慢性處方簽,第4季每月增加35%以上,在多重數據的支持下,顯示市場需求快速成長。
COPD龐大的市場也吸引多家公司進入,同年9月FDA核准Sanofi/Regeneron的Dupixent(dupilumab)擴充適應症至COPD,成為第一個被核准用於治療COPD的生物療法。GSK和Amgen/AstraZeneca亦分別以臨床數據驗證其氣喘藥品Nucala(mepolizumab)以及Tezspire(tezepelumab),可降低中度或重度COPD惡化的發生率。預期未來將有眾多競爭者進入市場,患者可望有更多治療選擇,而COPD的市場競爭也將更加熱鬧。
4. 代謝功能障礙相關脂肪性肝炎(MASH)治療藥品 Rezdiffra
Madrigal Pharmaceuticals 的Rezdiffra是一種甲狀腺激素受體β(Thyroid Hormone Receptor β, THRβ)促進劑,為第一個被批准用於MASH患者的治療方法,具有調節粒線體活化及脂肪分解、改善脂質代謝等效果。
在Rezdiffra未上市前,MASH多以Vitamin E和Pioglitazone治療,但治療效果不佳。Rezdiffra透過加速審查的途徑,以測量肝臟發炎和疤痕程度做為臨床試驗的替代終點,取得FDA的上市許可,但上市後Rezdiffra仍須進行後續的臨床研究,透過目前正在進行的54個月的研究來驗證Rezdiffra的臨床效益。根據Madrigal Pharmaceuticals的新聞稿,Rezdiffra每年藥費約為4.6萬美元,其在2024年的銷售額為1.77億美元。
Rezdiffra的上市對於後續針對MSAH治療的藥品開發者造成壓力,其治療效果需比Rezdiffra更好才有機會取得上市許可,但因MSAH的成因複雜,隨著對MASH致病機轉的了解,未來將有更多不同治療策略的藥品被開發以滿足不同的治療需求,且該市場龐大,因此,對於後續的開發者而言,仍是值得投入的市場。
5. 腫瘤浸潤淋巴細胞(TIL)療法 Amtagvi
Iovance Biotherapeutics的Amtagvi是一項個人化的TIL療法,2024年2月獲得CBER核准上市,可用於治療晚期黑色素瘤,這是繼嵌合抗原受體T細胞(Chimeric Antigen Receptor T, CAR-T)細胞療問世之後,免疫細胞療法跨入實體瘤市場的首例。根據臨床試驗的結果,Amtagvi在支持性總療效分析中,其客觀緩解率為31.4%,而目前的數據顯示,患者的中位數壽命為13.9個月,近半數的患者在4年後仍存活。根據Iovance Biotherapeutics的規劃,Amtagvi一次性治療的費用為51.5萬美元,相較於已上市的CAR-T細胞療法治療費用約在30~40萬美元之間,其治療費用較高。2024年前三季Amtagvi銷售額為5,490萬美元,隨著未來使用人數增加以及適應症擴增,預估銷售額持續看漲。
與目前的CAR-T細胞療法相同,TIL療法的細胞來源為患者,從患者的腫瘤細胞中收集TIL細胞,至體外擴增,之後再回輸至病患體內。這樣的治療方式,解決過往缺乏腫瘤特異性標記的問題,但仍有使用上的限制,例如TIL細胞取得不易,患者如不能進行手術或切除腫瘤細胞,則無法採用TIL療法,此外,即便取得患者的腫瘤細胞,自腫瘤細胞中純化TIL亦是一項困難的技術。因此,TIL療法雖然能解決目前T細胞療法的應用困難,其治療效果亦獲得藥證審查單位的認可,但仍需要突破多項技術以使TIL療法的可用性提升。
6. T細胞受體(TCR-T)細胞療法 Tecelra
Adaptimmune獲CBER核准Tecelra上市,可用於治療轉移性或不可切除滑膜肉瘤。而這項核准案,也讓Tecelra擁有許多個第一,包括第一個針對實體瘤的基因編輯細胞療法,第一個進入市場的TCR-T療法,也是十多年來針對滑膜肉瘤的第一個新療法。滑膜肉瘤為罕見惡性腫瘤,占所有軟組織肉瘤5~10%,5年存活率低於36%。Tecelra是自體免疫細胞療法,使用慢病毒載體(lentiviral vector)將基因編輯的T細胞受體基因送入細胞體內,修改T細胞的TCR,使其靶向並攻擊MHC I上帶有黑色素瘤相關抗原A4(MAGE-A4)的滑膜肉瘤。當Tecelra與MAGE-A4結合後,將會誘導T細胞增殖並分泌細胞因子,使癌細胞死亡,根據臨床試驗結果,Tecelra治療後,總體緩解率為43%,其中4.5%患者達到完成緩解,中位總生存期(Overall Survival, OS)約為17個月,七成的患者生存時間超過2年,相較於現有治療策略大有進展。
Tecelra治療前,患者須先接受MAGE-4A表達的伴同式診斷篩檢,確認表達情形再施予治療,屬於一次性的治療,費用為72.7萬美元。截至截稿前2025年1月,Adaptimmune Therapeutics尚未公布Tecelra的銷售結果,但根據2024年第3季公開的財報顯示,目前已有67%的商業保險計畫承保Tecelra,預估將帶動Tecelra的銷售成長。
與CAR-T細胞療法一樣,TCR-T療法皆是修改T細胞以辨識特定腫瘤抗原,但CAR-T細胞療法針對細胞表面抗原,而TCR-T則是針對MHC I呈現之細胞表面或細胞內抗原,因此,兩者在應用上產生差異。而兩種療法皆會產生細胞激素釋放症候群和神經毒性的副作用,但因TCR-T的作用機制更接近人體的免疫反應,因此理論上副作用可能較少,目前在Tecelra臨床試驗中沒有出現治療死亡事件,但仍需要更多研究驗證。
三、結語
從2024年美國FDA核准的新藥概況來看,FDA的審查流程持續支持創新藥品的開發,以滿足未被滿足的醫療需求。醫藥產業不斷加大對創新產品的研發投入,並透過多元技術途徑突破現有的技術瓶頸,推動醫療領域的持續創新。隨著科技進步,越來越多的創新藥品和治療技術應用於疾病的治療,並取得上市許可,為患者帶來了希望。儘管某些技術仍存在改進空間,但隨著多項技術突破的逐步彙集,我們有望在未來幾年見證療效進一步提升,並且迎來更多革命性的治療方法,這些方法將顯著改善患者的生活品質,甚至有可能重塑某些疾病的治療格局。
然而,高昂的價格、製造挑戰、商業策略及潛在的安全風險,仍然是新藥上市後需要密切關注的問題。為確保創新藥物能夠惠及更廣泛的患者群體,政策制定者、藥品開發者與保險機構需要彼此分工及合作,解決各部分可能面臨的問題,例如藥品供應、給付機制、或是安全監控技術及策略等,從而使這些創新治療能夠實現普及。